Es werden die Verfahren zur Anwendung von Wärmezyklen vor der Durchführung von Ölretentions- und Detonationstests bei Ammoniumnitrat mit hohem Stickstoffgehalt festgelegt.
2.1 Anwendungsbereich
Wärmezyklen vor Durchführung von Ölretentionsversuchen mit dem Düngemittel.
2.2 Prinzip und Definition
Die Probe wird in einem Erlenmeyerkolben von Raumtemperatur auf 50 °C erwärmt und rund zwei Stunden auf dieser Temperatur gehalten (Phase bei 50 °C). Anschließend wird sie auf 25 °C abgekühlt und zwei Stunden lang bei dieser Temperatur belassen (Phase bei 25 °C). Beide aufeinanderfolgenden Phasen (bei 50 °C und bei 25 °C) bilden zusammen einen Wärmezyklus. Nach Durchführung von zwei Wärmezyklen wird die Probe zur Bestimmung des Ölretentionsvermögens bei 20 ( ± 3) °C belassen.
2.3 Geräte
Übliches Laborgerät und insbesondere:
2.4 Durchführung
Eine Probenmenge von jeweils 70 (± 5) g wird in einen Erlenmeyerkolben gegeben, der dann verschlossen wird.
Der Kolben wird alle zwei Stunden vom 50 °C-Bad in das 25 °C-Bad und anschließend wieder in das 50 °C-Bad gestellt.
Die Temperatur der Bäder wird konstant gehalten und das Wasser mit rasch laufendem Rührer umgewälzt, um sicherzustellen, daß die Probe ganz untergetaucht ist.
Der Stopfen muß mit einem Schaumgummiüberzug vor Wasserdampfkondensation geschützt sein.
3.1 Anwendungsbereich
Wärmezyklen vor Durchführung von Detonationstests mit dem Düngemittel.
3.2 Prinzip und Definition
Die Probe wird in einem wasserdichten Behälter von Raumtemperatur auf 50 °C erwärmt und eine Stunde lang bei dieser Temperatur belassen (Phase bei 50 °C). Anschließend wird sie wieder auf 25 °C abgekühlt und eine Stunde lang bei dieser Temperatur belassen (Phase bei 25 °C). Die Kombination der beiden aufeinanderfolgenden Phasen bei 50 °C und 25 °C bildet einen Wärmezyklus. Nach Durchführung der angegebenen Zahl von Wärmezyklen wird die Probe bis zur Durchführung des Detonationsversuchs bei 20 (± 3) °C belassen.
3.3 Geräte
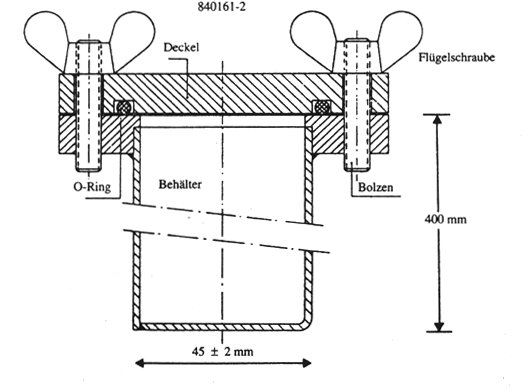
Abbildung 1
3.4 Durchführung
Eine für einen einzigen Detonationstest ausreichende Menge Düngemittel wird in den Stahlbehälter gegeben, der mit einem Deckel verschlossen wird.
Der Behälter wird in das Wasserbad gestellt, das Wasser auf 51 °C erwärmt und die Temperatur im Zentrum der Probe gemessen. Eine Stunde nach Erreichen von 50 °C wird die Kühlung eingeschaltet und das Wasser abgekühlt. Eine Stunde nach Erreichen der Temperatur von 25 °C im Zentrum der Probe ist die Heizung wieder anzustellen und der zweite Zyklus zu beginnen. Werden zwei Wasserbäder verwendet, so ist der Behälter nach jeder Abkühlung in das andere Wasserbad zu stellen.
Es wird eine Methode zur Bestimmung des Ölretentionsvermögens von Ammoniumnitrat-Einnährstoffdüngern mit hohem Stickstoffgehalt beschrieben.
Die Methode gilt für Prills und Granulate, die keine in Öl löslichen Stoffe enthalten.
Ölretention eines Düngemittels: Die Ölmenge, die vom Düngemittel zurückgehalten und unter festgelegten Betriebsbedingungen bestimmt und in Massen-% angegeben wird.
Eine Probe wird für eine bestimmte Dauer vollständig in Dieselöl getaucht, sodann läßt man das überschüssige Dieselöl unter genau festgelegten Bedingungen abtropfen. Man bestimmt die Massenzunahme der Probe (Teilmenge).
| Gasöl | |
| Viskosität max. | 5 mPas bei 40 °C, |
| Dichte: | 0,8 bis 0,85 g/ml bei 20 °C, |
| Schwefelgehalt: | 3/4 1,0 % (m/m), |
| Aschegehalt: | 3/4 0,1 % (m/m) |
Übliches Laborgerät und:
5.1 Waage mit einer Wägegenauigkeit von 0,01 g.
5.2 Bechergläser, Inhalt 500 ml.
5.3 Trichter aus Kunststoff, vorzugsweise mit einer zylindrischen Wandung am oberen Ende, Durchmesser ca. 200 mm.
5.4 Prüfsieb, Maschenweite 0,5 mm, das auf den Trichter (5.3) aufgesetzt werden kann.
Anmerkung: Die Abmessungen des Trichters und des Prüfsiebes müssen so gewählt werden, daß nur wenige Körner übereinanderliegen und das Öl leicht abfließen kann.
5.5 Papierfilter, schnellfiltrierend, weich (Krepp), Flächendichte 150 g/m2.
5.6 Saugfähiger Zellstoff (Labortücher, saugkräftig).
6.0 Mit derselben Probe werden rasch hintereinander zwei Einzelbestimmungen durchgeführt.
6.1 Mit dem Prüfsieb (5.4) werden Teilchen mit weniger als 0,5 mm Durchmesser entfernt. Für eine Einzelbestimmung werden 50 g Probe auf 0,01 g genau abgewogen und in das Becherglas (5.2) gegeben. Ausreichend Dieselöl (Punkt 4) zugeben, bis die Prills vollständig bedeckt sind, und sorgfältig umrühren, um sicherzustellen, daß die Oberflächen sämtlicher Prills vollständig benetzt sind. Becher mit einem Uhrglas abdecken und eine Stunde bei 25 ( ± 2) °C stehenlassen.
6.2 Der gesamte Inhalt des Becherglases wird durch den mit einem Prüfsieb (5.4) versehenen Trichter (5.3) gefiltert. Die im Sieb zurückgehaltene Probe eine Stunde lang abtropfen lassen, damit das überschüssige Dieselöl möglichst vollständig abfließen kann.
6.3 Zwei Lagen Filterpapier (5.5) (etwa 500 x 500 mm) übereinander auf eine glatte Oberfläche legen, die 4 Seiten der beiden Filterpapiere so nach oben falten, daß ein etwa 4 cm breiter Randstreifen entsteht und die Prills nicht fortrollen können. Man lege in die Mitte der Filterpapiere zwei Lagen eines saugfähigen Labortuchs (5.6), schütte den gesamten Inhalt des Filters (5.4) darauf und verteile diesen gleichmäßig mit einer weichen, flachen Bürste. Nach zwei Minuten hebe man eine Seite des saugfähigen Labortuchs an, befördere die Prills auf die darunterliegenden Filterpapiere und verteile sie gleichmäßig mit einer Bürste.
Eine weitere Filterpapierlage mit ebenfalls nach oben gefalteten Randstreifen auf die Probe legen und die Prills zwischen den Filterpapieren mit kreisförmigen Bewegungen und unter leichtem Druck rollen. Nach jeweils acht kreisförmigen Bewegungen die gegenüberliegenden Seiten der Filterpapiere anheben und die an die Ränder gerollten Prills wieder in die Mitte bringen. Dabei ist folgendermaßen vorzugehen: Jeweils vier volle Kreisbewegungen im und gegen den Uhrzeigersinn, danach werden die Prills wie vorstehend beschrieben in die Mitte zurückgerollt. Dieses Verfahren wird jeweils dreimal durchgeführt (24 x Kreisbewegungen, 2 x Anheben der Kanten). Danach schiebe man einen neuen Filterbogen vorsichtig zwischen den zuunterst liegenden und den darüberliegenden Bogen und lasse die Prills durch Anheben der seitlichen Kanten des letztgenannten Bogens auf den neuen Bogen abrollen. Nach Bedecken der Prills mit einem neuen Filterbogen wird der oben beschriebene Abrollvorgang wiederholt. Unmittelbar nachher werden die Prills in eine austarierte Schale geschüttet und durch Rückwägung das Gewicht der zurückgehaltenen Menge an Dieselöl auf 0,01 g genau ermittelt.
6.4 Wiederholung des Abrollvorgangs und Rückwägung
Beträgt die in der Teilmenge enthaltene Menge Dieselöl mehr als 2 g, so wird diese auf einen frischen Satz Filterpapierbögen gegeben, anschließend wird ein neuer Abrollvorgang mit Anheben der Ecken entsprechend Abschnitt 6.3 (2 x 8 Kreisbewegungen, dazwischen einmal Anheben) durchgeführt. Danach wird die Teilmenge erneut gewogen.
7.1 Berechnungsverfahren und Gleichung
Die Ölretention, ausgedrückt als Prozentsatz bezogen auf die Masse der abgesiebten Teilmenge, wird nach folgender Formel berechnet:
![]()
wobei
m1 = Masse der abgesiebten Teilmenge (6.1) in Gramm;
m2 = Masse der Teilmenge nach 6.3 beziehungsweise 6.4 Ergebnis der letzten Rückwägung in Gramm.
Als Ergebnis gilt das arithmetische Mittel der beiden Einzelbestimmungen.
Es wird ein Verfahren zur Bestimmung des Gehaltes an brennbaren Stoffen in Ammoniumnitrat-Einnährstoffdüngern mit hohem Stickstoffgehalt festgelegt.
Das aus anorganischem Füllstoff entstehende Kohlendioxid wird vor der Bestimmung mit einer Säure ausgetrieben. Die organischen Verbindungen werden mit Hilfe einer Chromschwefelsäuremischung oxydiert. Das entstehende Kohlendioxid wird in einer Bariumhydroxidlösung absorbiert. Der Niederschlag wird in eingestellter Salzsäurelösung aufgelöst und durch Rücktitration mit einer Natriumhydroxidlösung bestimmt.
3.1 Chrom(VI)-oxid, CrO3, analysenrein.
3.2 Schwefelsäure, auf 60 Volumenprozent verdünnt:
3.3 Silbernitrat: Lösung 0,1 M.
3.4 Bariumhydroxid:
3.5 Salzsäure: Standardlösung 0,1 M.
3.6 Natriumhydroxid: Standardlösung 0,1 M.
3.7 Bromphenolblau: Lösung von 0,4 g/l in Wasser.
3.8 Phenolphthalein: Lösung von 2 g/l in Äthanol zu 60 Volumenprozent.
3.9 Stickstoff, frei von Kohlenstoffverbindungen, insbesondere frei von CO und CO2, alternativ: Stickstoff, technisch und Adsorptionsmasse Natriumhydroxid auf Träger.
3.10 Entmineralisiertes Wasser, das zur Austreibung des CO2 kurz vorher zum Sieden gebracht wird.
4.1 Übliches Laborgerät, insbesondere:
4.2 Geräte mit folgenden Komponenten-Verbindungen, wenn möglich mit kugelförmigen Schliffstopfen (siehe Abbildung 2).
4.2.1 Reaktionskolben B von 500 ml, mit seitlichem Hals und rundem Boden.
4.2.2 Vigreux-Fraktionieraufsatz, Länge zirka 150 mm (C').
4.2.3 Intensivkühler C, mit Oberflächenverdopplung, Länge 200 mm.
4.2.4 Drechselflasche D zum Auffangen von eventuell überdestillierter Säure.
4.2.5 Eisbad E zur Abkühlung der Drechselflasche.
4.2.6 Zwei Absorber F1 und F2, Durchmesser 32 bis 35 mm, deren Gasverteiler aus einer 10 mm-Scheibe aus Glasfrittenmaterial mit niedriger Porosität besteht.
4.2.7a Bei Verwendung von Reinstickstoff
Stickstoff-Druckgasflasche mit Druckreduzierventil und Ventil zur Regelung des Volumenstromes.
4.2.7b Bei Verwendung von technischem Stickstoff
Absorber, gefüllt mit Natriumhydroxid auf Träger zwischen Glaswollepackungen zur Reinigung des Stickstoffs von Kohlendioxid am Gaseinlaß der Apparatur.
4.2.8 Saugpumpe und Saugkraftregler G, bestehend aus einem in das Ableitungsrohr eingefügten T-förmigen Glasstück, dessen freier Arm mit einem kurzen, mit einer Schraubverbindung ausgestatteten Kautschukschlauch an ein feines Kapillarrohr angeschlossen ist.
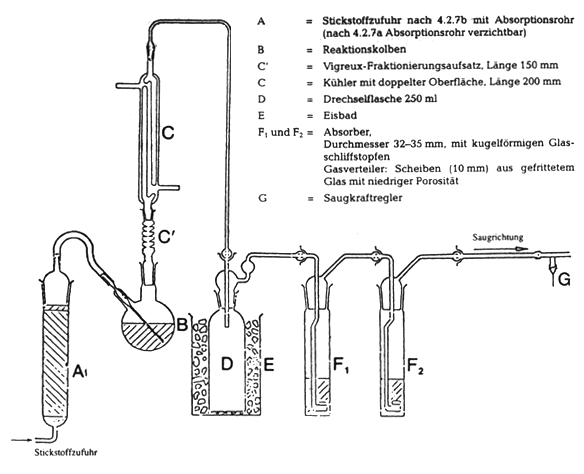
Abbildung 2
Rund 10 g auf 0,001 g genau abgewogenes Ammoniumnitrat.
Probe in den Reaktionskolben B einfüllen. 100 ml H2SO4 (3.2) zugeben. Bei Raumtemperatur lösen sich die Prills oder das Granulat in etwa 10 Minuten auf.
Aufbau des Geräts nach Schema: Die Stickstoffquelle (4.2.7) mittels Schlauch über ein Rückschlagventil (5 bis 6 mm Quecksilbersäule) an das in den Reaktionskolben eintauchende Zuführrohr anschließen. Einbau des Vigreux-Fraktionsaufsatzes (C') und des an das Kühlwasser angeschlossenen Kühlers (C). Nach Einstellung des Stickstoffstroms auf leichtes Durchströmen der Lösung wird diese auf den Siedepunkt erwärmt und 2 Minuten auf dieser Temperatur gehalten. Danach sollten sich keine Bläschen mehr bilden. Bei Fortsetzung der Bläschenbildung wird die Erwärmung 30 Minuten fortgesetzt. Anschließend Lösung mindestens 20 Minuten lang im Stickstoffstrom abkühlen lassen.
Gerät nach Schema fertig zusammenbauen. Kühler mit Drechselflasche (D) verbinden und diese an die Absorber F1 und F2 anschließen. Während des Zusammenbaus muß Stickstoff strömen.
Rasch 50 ml Bariumhydroxidlösung (3.4) in jeden Absorber (F1 und F2) einfüllen.
Stickstoff etwa 10 Minuten durchströmen lassen. Die Lösung in den Absorbern muß klar bleiben. Andernfalls ist das Karbonatbeseitigungsverfahren zu wiederholen.
Nach Zurückziehen des Stickstoffzufuhrrohrs werden durch den Seitenarm des Reaktionskolbens (B) rasch 20 g Chrom(VI)-oxid (3.1) und 6 ml Silbernitratlösung (3.3) eingefüllt.
Vorsicht!
Der Einsatz siedender Chromsäure unter vermindertem Druck ist gefährlich und erfordert entsprechende Vorsichtsmaßnahmen.
Das Gerät wird an die Saugpumpe angeschlossen und der Stickstoffstrom so geregelt, daß die Sinterglas-Absorber F1 und F2 ständig von Gasblasen durchflossen werden.
Inhalt des Reaktionskolbens (B) 90 Minuten sieden lassen*). Gegebenenfalls muß die Saugpumpe in Gang gesetzt werden, da die Scheiben während des Tests durch den Bariumkarbonatniederschlag verstopft werden können. Der Vorgang ist zufriedenstellend, wenn die Bariumhydroxidlösung im Absorber F2 klar bleibt. Andernfalls ist er zu wiederholen. Heizung ausschalten und Gerät auseinandernehmen. Zur Entfernung des Bariumhydroxids beide Gasverteiler mit frisch abgekochtem, destilliertem Wasser (3.10) innen und außen reinigen und das Waschwasser im entsprechenden Absorber auffangen. Die Verteiler nacheinander in ein 600-ml-Becherglas legen, das später zur Bestimmung verwendet wird.
Den Inhalt des Absorbers F2 und anschließend des Absorbers F1 rasch durch den Glasfiltertiegel im Vakuum filtrieren. Den Niederschlag mit Waschwasser (3.10) der Absorber spülen und den Tiegel mit 50 ml Wasser gleicher Qualität waschen. Tiegel in das 600-ml-Becherglas stellen und etwa 100 ml Wasser (3.10) zugeben. In beide Absorber 50 ml Wasser einfüllen und einen Stickstoffstrom 5 Minuten lang durch die Verteiler fließen lassen. Die einzelnen Wassermengen zu dem Wasser im Becherglas geben und den Vorgang wiederholen, um sicherzustellen, daß die Verteiler gut gespült werden.
Fünf Tropfen Phenolphthalein (3.8) in das Becherglas geben. Die Lösung wird rot. Anschließend tropfenweise Salzsäure (3.5) zugeben, bis die Färbung vollständig verschwindet.
Die Lösung im Tiegel gut schütteln, um sicherzustellen, daß sich die Rotfärbung nicht wieder einstellt. Fünf Tropfen Bromphenolblau zugeben und mit Salzsäure bis zur Gelbfärbung titrieren. Nochmals 10 ml Salzsäure zugeben.
Die Lösung bis zum Siedepunkt erwärmen und nicht länger als eine Minute sieden lassen. Genau prüfen, daß die Flüssigkeit keinen Niederschlag mehr enthält.
Abkühlen lassen und mit Natriumhydroxidlösung (3.6) zurücktitrieren.
Parallel zur Bestimmung ist ein Blindversuch mit der gleichen Arbeitsmethode und den gleichen Reagenzienmengen durchzuführen.
Der Gehalt an organischen Verbindungen (C), dargestellt in Prozent der gesamten Kohlenstoffmasse, wird nach folgender Formel berechnet:
![]()
Hierbei sind:
| E | Masse der entnommenen Probe in Gramm; |
| V1 | Gesamtvolumen der nach dem Phenolphthalein-Farbumschlag hinzugefügten 0,1 M Salzsäure in ml; |
| V2 | Volumen der 0,1 M Natriumhydroxidlösung in ml für die Rücktitration: |
Diese Methode dient der Bestimmung des pH-Wertes einer Lösung von Ammoniumnitratdünger mit hohem Stickstoffgehalt.
Messung des pH-Wertes einer Ammoniumnitratlösung mit einem pH-Meßgerät.
Destilliertes oder entmineralisiertes und kohlendioxidfreies Wasser.
Man löst 3,40 (± 0,01) g Kaliumdihydrogenorthophosphat (KH2PO4) in etwa 400 ml Wasser auf. Dann löst man 3,55 (± 0,01) g Natriumhydrogenorthophosphat (Na2HPO4) in etwa 400 ml Wasser auf. Man gibt die beiden Lösungen quantitativ in einen Meßkolben von 1 000 ml, füllt bis zur Marke auf und mischt. Diese Lösung wird in einem luftdicht verschlossenen Gefäß aufbewahrt.
Man löst 10,21 (± 0,01) g Kaliumhydrogenphthalat (KHC8O4H4) in Wasser auf, gießt die Lösung quantitativ in einen Meßkolben von 1 000 ml um, füllt bis zur Marke auf und mischt.
Diese Lösung ist in einem luftdicht verschlossenen Behälter aufzubewahren.
3.3 Es können gebrauchsfertige, handelsübliche Pufferlösungen verwendet werden.
pH-Meßgeräte mit Glas-, Kalomel- oder entsprechenden Elektroden und einer Empfindlichkeit von 0,05 pH-Einheiten.
Das pH-Meßgerät (4) ist bei einer Temperatur von 20 (± 1) °C unter Verwendung der Pufferlösungen (3.1, 3.2 oder 3.3) zu eichen. Man leitet während des gesamten Versuchs einen leichten Stickstoffstrom über die Oberfläche der Lösung.
10 (± 0,01) g Probe sind in 100 ml Wasser in einem 250-ml-Becherglas zu lösen. Nichtlösliche Bestandteile sind durch Filtrieren, Dekantieren oder Zentrifugieren zu entfernen.
Der pH-Wert der klaren Lösung wird bei einer Temperatur von 20 ( ± 1) °C nach dem zur Eichung des Meßgeräts angewandten Verfahren gemessen.
Die Ergebnisse sind in pH-Einheiten mit einer Fehlergrenze von 0,1 Einheiten und der gegebenen Temperatur anzugeben.
Diese Methode dient der Festlegung eines Verfahrens zur Bestimmung der Korngröße von Ammoniumnitrat-Einnährstoffdüngern mit hohem Stickstoffgehalt.
Eine Probe wird von Hand oder mechanisch durch einen Satz von drei Sieben gesiebt. Der Rückstand auf jedem Sieb wird ausgewogen. Die relativen Anteile der die vorgeschriebenen Siebe passierenden Probemenge werden berechnet.
3.1 Standardisierte Prüfsiebe aus Drahtgewebe mit 200 mm Durchmesser und Maschenweite von 2,00 mm, 1,00 mm und 0,5 mm mit zugehörigem Deckel und Auffanggefäß.
3.2 Waage mit einer Wägegenauigkeit von 0,1 g.
3.3 Mechanische Schüttelvorrichtung, falls vorhanden, die die Probemenge sowohl in vertikaler als auch in horizontaler Richtung bewegt.
4.1 Die Probe wird in repräsentative Teilmengen von rund 100 g unterteilt.
4.2 Diese Teilmengen werden auf 0,1 g genau gewogen.
4.3 Der Siebsatz ist in aufsteigender Reihenfolge anzuordnen (Auffanggefäß, 0,5 mm, 1 mm, 2 mm). Die abgewogene Probe wird auf das oberste Sieb gebracht, das mit dem Deckel verschlossen wird.
4.4 Man schüttelt von Hand oder mechanisch und zwar so, daß sowohl vertikale als auch horizontale Bewegungen ausgeführt werden; schüttelt man von Hand, so klopft man gelegentlich auf die Siebe. Man schüttelt zehn Minuten oder bis der Siebdurchsatz weniger als 0,1 g/Min. beträgt.
4.5 Die Siebe werden nacheinander abgenommen. Der Siebrückstand wird entnommen. Gegebenenfalls wird das entsprechende Sieb von der Gegenseite her mit einem weichen Pinsel leicht ausgepinselt.
4.6 Man wiegt den Rückstand von den einzelnen Sieben und vom Auffanggefäß auf 0,1 g genau aus.
5.1 Die Massenanteile sind in % der Summe der Massenanteile (und nicht der ursprünglichen Einwaage) umzurechnen.
Der prozentuale Anteil im Auffanggefäß (d. h. Korngröße < 0,5 mm) ist als A % zu berechnen. Der Anteil des Rückstandes auf dem 0,5-mm-Sieb ist als B % zu berechnen. Der das 1,00-mm-Sieb passierende Anteil ist als (A + B) % zu berechnen.
Die Summe der Massenanteile sollte um höchstens 2 % von der ursprünglichen Einwaage abweichen.
5.2 Es sind mindestens zwei getrennte Bestimmungen durchzuführen. Die einzelnen Ergebnisse für A dürfen nicht um mehr als 1,0 % absolut und diejenigen für B nicht um mehr als 1,5 % absolut voneinander abweichen. Falls dies nicht der Fall ist, ist der Test zu wiederholen.
Für die beiden Werte A und A + B ist der Durchschnittswert anzugeben.
Es ist ein Verfahren zur Bestimmung des Gehalts an Chlor (Chloridionen) in Ammoniumnitrat-Einnährstoffdüngern mit hohem Stickstoffgehalt festgelegt.
Die in Wasser gelösten Chloridionen werden in saurem Milieu durch eine potentiometrische Titration mit Silbernitrat-Maßlösung bestimmt.
Destilliertes oder vollständig entmineralisiertes Wasser, frei von Chlorid.
3.1 Azeton, analysenrein.
3.2 Reine konzentrierte Salpetersäure, Dichte bei 20 °C = 1,40 g/ml.
3.3 Silbernitrat-Maßlösung 0,1 M; in brauner Glasflasche aufbewahren.
3.4 Silbernitrat-Maßlösung 0,004 M; zum Zeitpunkt der Verwendung herstellen.
3.5 Kaliumchlorid-Standardlösung 0,1 M; 3,7276 g analysenreines Kaliumchlorid, das zuvor eine Stunde bei 130 °C getrocknet und im Exsikkator auf Raumtemperatur abgekühlt worden ist, werden auf 0,1 mg genau gewogen, in Wasser gelöst und quantitativ in einen 500-ml-Meßkolben umgegossen; der Kolben wird bis zur Marke aufgefüllt und die Lösung durchgemischt.
3.6 Kaliumchlorid-Standardlösung 0,004 M; zum Zeitpunkt der Verwendung herzustellen.
4.1 Potentiometer mit Silberelektrode und Kalomel-Bezugselektrode: Empfindlichkeit 2 mV. Meßbereich von - 500 bis + 500 mV.
4.2 Brücke, die die gesättigte Kaliumnitratlösung enthält und mit der Kalomelelektrode (4.1) verbunden wird. Die Brücke ist an den Enden mit porösen Stopfen versehen.
Anmerkung: Diese Brücke ist nicht erforderlich, wenn eine Silberelektrode und eine Quecksilber(1)-Sulfatelektrode als Bezugselektrode verwendet wird.
4.3 Magnetrührer mit einem teflonbeschichteten Rührstäbchen.
4.4 Mikrobürette mit Feindosierventil und 0,01-ml-Graduierung.
5.1 Einstellung des Titers der Silbernitratlösungen
(1) 5,00 ml und 10,00 ml der entsprechenden Kaliumchlorid-Standardlösung (3.6) werden in zwei niedrige Bechergläser mit geeignetem Fassungsvermögen (z.B. 250 ml) gegeben. Die Titration des Inhalts jedes Bechers wird folgendermaßen durchgeführt:
(2) 5 ml Salpetersäure (3.2), 120 ml Azeton (3.1) hinzufügen und das Gesamtvolumen mit Wasser auf ca. 150 ml auffüllen. Rührstäbchen des Magnetrührers (4.3) in den Titrationsbecher einführen und Rührgerät einschalten. Silberelektrode (4.1) und das freie Ende der Brücke (4.2) in die Lösung eintauchen.
Die Elektroden an das Potentiometer (4.1) anschließen und nach Nullabgleich den Wert des Ausgangspotentials des Gerätes notieren.
(3) Man titriert, indem mit der Mikrobürette (4.4) entsprechend der angewandten Kaliumchlorid-Standardlösung anfänglich 4 bzw. 9 ml Silbernitratmaßlösung hinzugegeben werden. Die Zugabe der 0,004-M-Titerlösung wird in Teilmengen von 0,1 ml und der 0,1-M-Titerlösung in Teilmengen von 0,05 ml fortgesetzt. Nach jeder Zugabe ist die Potentialeinstellung abzuwarten.
(4) In den beiden ersten Spalten einer Tabelle sind die zugefügten Volumina und die entsprechenden Potentialwerte zu notieren.
(5) In einer dritten Spalte der Tabelle werden die sukzessiven Potentialzunahmen (Δ1E) notiert. In einer vierten Spalte notiert man dann die positiven oder negativen Unterschiede (Δ2E) zwischen den Potentialdifferenzen (Δ1E). Das Ende der Titration wird mit der Zugabe der Teilmenge von 0,1 bzw. 0,05 ml (V1) Silbernitratlösung erreicht, die den Höchstwert von Δ1E ergibt.
(6) Das genaue Volumen (Veq) der Silbernitratlösung, die dem Reaktionsendpunkt entspricht, erhält man durch folgende Formel:
| Veq | = | V0 + (V1 * b/B) wobei: |
| V0 | = | Gesamtvolumen der Silbernitratlösung unmittelbar unterhalb des Volumens, das den höchsten Zuwachs Δ1E ergibt, in ml; |
| V1 | = | Volumen der letzten hinzugefügten Teilmenge der Silbernitratlösung (0,1 oder 0,05 ml) in ml; |
| b | = | Wert des letzten positiven Δ2E; |
| B | = | Summe der absoluten Werte des letzten positiven Δ2E und des ersten negativen Δ2E (siehe Beispiel in Tabelle 1). |
Tabelle 1 Beispiel
Silbernitratlösung V |
E |
||
4,90 5,00 5,10 5,20 |
211 283 306 319 |
35 72 23 13 |
+ 37 - 49 - 10 |
5.2 Blindversuch
Man führt einen Blindversuch durch und berücksichtigt diesen bei der Berechnung des Endergebnisses.
Das Ergebnis des Reagenzienblindwertes V4, in ml, wird nach folgender Formel erhalten:
V4 = 2V3 - V2
wobei:
| V2 = | genaues Volumen Veq der Silbernitratlösung, die der Titration von 10 ml der verwendeten Kaliumchlorid-Standardlösung entspricht, in ml; |
| V3 = | genaues Volumen Veq der Silbernitratlösung, die der Titration von 5 ml der verwendeten Kaliumchlorid-Standardlösung entspricht, in ml. |
5.3 Kontrollbestimmung
Der Blindversuch dient gleichzeitig dazu, das einwandfreie Funktionieren des Gerätes und die korrekte Durchführung des Testverfahrens zu prüfen.
5.4 Bestimmung
10 bis 20 g der Probe werden auf 0,01 genau abgewogen und quantitativ in ein 250-ml-Becherglas gegeben.
Zur eingewogenen Teilmenge fügt man 20 ml Wasser, 5 ml Salpetersäure (3.2) und 120 ml Azeton (3.1) zu und füllt mit Wasser auf ca. 150 ml auf. Rührstab des Magnetrührers (4.3) in das Becherglas einführen, dieses auf das Rührgerät stellen und das Rührgerät einschalten.
Die Silberelektrode (4.1) und das freie Ende der Brücke (4.2) in die Lösung einführen, die Elektroden an das Potentiometer (4.1) anschließen und den Wert des Ausgangspotentials nach Prüfung des Nullstandes des Gerätes notieren. Titrieren, indem mit der Mikrobürette (4.4) die Silbernitratlösung in Teilmengen von 0,1 ml hinzugefügt wird. Nach jeder Hinzugabe Stabilisierung des Potentials abwarten.
Die Titrierung gemäß 5.1 fortsetzen, wobei ab Absatz 4 zu beginnen ist.
Das Analyseergebnis ist in Prozent Chlor des zur Untersuchung eingereichten Düngemittels anzugeben.
Man berechnet den Gehalt an Chlor (Cl) nach folgender Formel:
![]()
| T = | Molarität der verwendeten Silbernitratlösung; |
| V4 = | Ergebnis des Blindversuchs in ml (5.2); |
| V5 = | Wert von Veq in ml entsprechend der Bestimmung (5.4); |
| m = | Masse der Teilmenge in g. |
Diese Methode dient der Bestimmung von Kupfer in Ammoniumnitrat-Einnährstoffdüngern mit hohem Stickstoffgehalt.
Die Probe wird in verdünnter Salzsäure gelöst. Die Lösung wird verdünnt und der Kupfergehalt durch Atomabsorptionsspektrophotometrie bestimmt.
3.1 Salzsäure (Dichte bei 20 °C = 1,18 g/ml).
3.2 Salzsäure, 6 M.
3.3 Salzsäure, 0,5 M.
3.4 Ammoniumnitrat.
3.5 Wasserstoffperoxid, 30 %ig.
3.6 Kupferlösung1) (Stammlösung): 1 g reines Kupfer auf 0,001 g genau abwiegen, in 25 ml 6 M Salzsäure (3.2) auflösen, portionenweise 5 ml Wasserstoffperoxid (3.5) hinzugeben und mit Wasser auf 1l auffüllen, 1 ml dieser Lösung enthält 1 000 μg Kupfer (Cu).
3.6.1 Kupferlösung (verdünnt): 10 ml Stammlösung (3.6) mit Wasser auf 100 ml auffüllen und 10 ml der so erhaltenen Lösung mit Wasser auf 100 ml auffüllen, 1 ml der zuletzt erhaltenen Lösung enthält 10 μg Kupfer.
Diese Lösung ist zum Zeitpunkt ihrer Verwendung herzustellen.
Atomabsorptionsspektrophotometer mit Kupferlampe (324,8 nm).
25 g der Probe werden auf 0,001 g genau in ein 400-ml-Becherglas abgewogen. Man gibt vorsichtig 20 ml Salzsäure (3.1) zu. (Durch die Bildung von Kohlendioxid kann es zu einer heftigen Reaktion kommen). Falls erforderlich, ist weitere Salzsäure zuzugeben.
Nach Beendigung der Gasentwicklung wird die Lösung unter gelegentlichem Rühren mit einem Glasstab in einem Wasserbad bis zur Trockne eingedampft. Dann fügt man 120 ml Wasser und 15 ml 6 M Salzsäure (3.2) zu. Mit dem Glasstab, der im Becherglas verbleiben sollte, wird umgerührt. Das Becherglas wird mit einem Uhrglas abgedeckt. Durch vorsichtiges Kochen wird der Rückstand völlig gelöst. Anschließend wird abgekühlt.
Unter Ausspülen des Becherglases mit 5 ml 6 M Salzsäure (3.2) und zweimaligem Nachspülen mit 5 ml kochendem Wasser wird die Lösung quantitativ in einen 250-ml-Meßkolben überführt. Man füllt bis zur Marke mit 0,5 M Salzsäure (3.3) auf und mischt sorgfältig.
Man filtriert durch ein kupferfreies Filterpapier ab; die ersten 50 ml sind zu verwerfen.
Eine Blindprobenlösung, zu der keine Probe hinzugefügt wird, ist herzustellen und bei der Berechnung der Endergebnisse zu berücksichtigen.
5.3.1 Zubereitung der Probe und des Lösungen für den Blindversuch
Die Probenlösung (5.1) und die Blindprobenlösung (5.2) wird mit 0,5 M Salzsäure (3.3) auf eine für den Meßbereich des Spektrophotometers optimale Konzentration verdünnt. Für gewöhnlich ist keine Verdünnung erforderlich.
5.3.2 Herstellung der Kalibrationslösung
Durch Verdünnung der Standardlösung (3.6.1) mit 0,5 M Salzsäure (3.3) werden mindestens 5 Lösungen hergestellt, die dem optimalen Meßbereich des Spektrophotometers 0 bis 5,0 μg/l Cu entsprechen.
Vor dem Auffüllen bis zur Marke wird jeder Lösung Ammoniumnitrat (3.4) zugegeben, um eine Endkonzentration von 100 mg/ml zu erhalten.
Das Spektrophotometer (4) wird auf eine Wellenlänge von 324,8 nm eingestellt. Man verwendet zur Messung eine oxydierende Luft-Acetylenflamme. Nacheinander werden die Lösungen (5.3.2), die Probe sowie die Blindprobenlösung (5.3.1) dreifach eingesprüht. Das Gerät wird zwischen jedem Meßvorgang mit destilliertem Wasser durchgespült. Zur Erstellung der Kurve werden die durchschnittlichen Extinktionswerte jeder Maßlösung auf der Ordinate und die entsprechenden Kupferkonzentrationen in μg/ml auf der Abszisse aufgetragen.
Die Kupferkonzentration der Proben- und Blindprobenlösung wird mit Hilfe der Kurve bestimmt.
Der Kupfergehalt der Probe wird unter Berücksichtigung der Einwaage, der im Verlauf der Analyse durchgeführten Verdünnungen und des Blindwerts berechnet. Das Ergebnis wird in mg Cu/kg angegeben.